Valine
Alpha-keto gamma-monamino monocarboxylic acid with a branched carbon atom chain (a-aminoisovaleric acid; 2-amino-3-methylbutanoic acid). Valine is one of the ten amino acids most commonly found on Earth.

Three amino acids have a branched chain – leucine, valine, and isoleucine.
These are essential amino acids. The metabolism of leucine, valine, and isoleucine occurs most intensively not in the liver, like many amino acids, but in muscle and adipose tissue, kidneys, and brain. The initial breakdown reactions of all three amino acids are identical, for example, valine:

Initially, branched-chain amino acids in muscle and other organs (not the liver) are transaminated with alpha-ketoglutarate, catalyzed by branched-chain aminotransferases (which are not present in the liver). The resulting alpha-keto acids are transported to the liver. Here, their oxidative decarboxylation takes place, catalyzed by the specific branched-chain alpha-keto acid dehydrogenase (mitochondrial poliferm complex with decarboxylase, transacylase, and dihydrolipoyl dehydrogenase activity). Phosphorylation and dephosphorylation of the dehydrogenase complex regulate the metabolism of branched-chain amino acids.
After decarboxylation, the further breakdown reactions of all three amino acids differ. Valine is a purely glucogenic amino acid, isoleucine is both glucogenic and ketogenic, while leucine is purely ketogenic. Leucine breaks down to acetoacetate and acetyl-CoA via 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). The final breakdown reactions of valine and isoleucine coincide with the oxidation of odd-chain fatty acids and the breakdown of threonine and methionine – propionyl-CoA is formed, followed by the isomerization of methylmalonyl-CoA to succinyl-CoA.
The isomerization is catalyzed by methylmalonyl-CoA mutase, whose coenzyme is 5-deoxyadenosylcobalamin (a derivative of vitamin B12). In the absence of cobalamin (vitamin B12), the activity of this mutase decreases.
Pathway of valine, isoleucine, and leucine metabolism
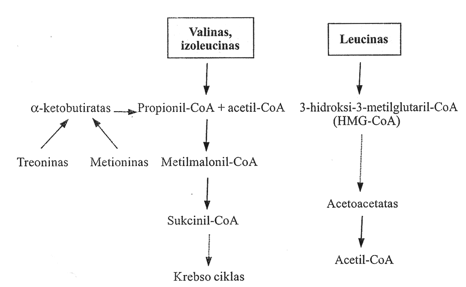
During leucine catabolism, acetyl-CoA and acetoacetate are formed from 3-hydroxy-3-methylglutaryl-CoA.
This reaction occurs in the mitochondria of mammalian liver, kidney, and heart cells. This explains the strong ketogenic effect of leucine.
There are several metabolic disorders of branched-chain amino acids known, but the best studied is maple syrup urine disease. It occurs in 1 in 200,000 newborns. Other disorders are even rarer. Maple syrup urine disease (leucinosis) is an inherited disorder; it arises from a genetic defect of branched-chain alpha-keto acid dehydrogenase, disrupting the oxidative decarboxylation of branched-chain alpha-keto acids. If this enzyme is completely inactive, mental retardation, acidosis, and lethargy can occur. The disease is characterized by a specific urine odor resembling maple syrup or burnt sugar. There is an increased level of branched-chain amino acids and corresponding alpha-keto acids in the blood plasma and urine. Therefore, this disease is sometimes also called branched-chain ketoaciduria. Additionally, branched-chain alpha-hydroxy acids are found in the urine, formed by reducing alpha-keto acids. Symptoms of the disease appear at the end of the first week after birth; it starts with vomiting, sometimes lethargy. Untreated infants die within the first year of life. Patients are given a diet consisting of an amino acid mixture without leucine, valine, and isoleucine. Once the levels of these amino acids in the blood decrease to normal concentrations, branched-chain amino acids (usually with milk) are added to the diet, but only in amounts necessary for protein biosynthesis. If treatment is started within the first week after birth, the course of the disease can be mitigated. Very high doses of vitamin B1 (thiamine) help in treatment. In the case of methylmalonic aciduria (occurring in 1 in 10,000), it is necessary to reduce the intake of valine, isoleucine, threonine, and methionine in the diet (amino acids that form propionyl-CoA). This disease arises from a genetic defect of methylmalonyl-CoA mutase, dependent on insufficient vitamin B12 (adenosylcobalamin) (vitamin B12 absorption may be impaired, or vitamin B12 cannot convert to a coenzyme). Methylmalonic acid is found in the urine of patients with pernicious anemia.
Source | Glossary of Most Commonly Used Biomedical Terms and Concepts | Lithuanian University of Health Sciences | Academician Professor Antanas Praškevičius, Professor Laima Ivanovienė